
Record-Scale AI Screening with Model Medicines on Google Cloud: GALILEO™ Achieves 325 Billion Molecule Throughput for Oncology Drug Discovery
Insights

Opening the Door to Trillion-Scale Drug Discovery on Google Cloud

Tyler Umansky
Head of Platform and Machine Learning
Abstract
Modern drug discovery pipelines chase ever-larger datasets and model sizes, yet most production stacks still top out at screening only millions to low billions of candidates. We argue that throughput is now the primary bottleneck in AI Drug Discovery, not training data availability. Current approaches falter in two ways: (i) model extrapolation degrades as rare chemotypes are swamped by million to billion scale training data, encouraging memorization; and (ii) computational cost explodes as architectures scale, making large scale screening impractical. We address both with an architecture that (a) trains for out-of-distribution generalization using small, deliberately diverse datasets, (b) prioritizes lightweight, parametric models capable of efficient inference, all engineered end-to-end on Google Cloud infrastructure. Our AI-drug discovery engine, GALILEO™, achieved 325 billion molecule throughput, the first to reach the hundred-billion scale. This campaign delivered MDL-4102, a first-in-class, selective BRD4 inhibitor with no observable BRD2/BRD3 activity, with chemical novelty versus clinical BET inhibitors (based on ECFP4 Tanimoto). Alongside MDL-4102, GALILEO™ previously enabled the discovery of MDL-001, a category-defining therapeutic in the virology space. By pairing models that generalize with throughput engineering, GALILEO™ shows that novel therapeutic discovery scales with compute.
Background
The universe of possible drug-like molecules is unimaginably vast (10^60), so large that the total annual output of all the world’s laboratories barely scratches its surface. This scale has motivated virtual screening efforts to expand from testing millions to billions of candidate molecules in silico. With more candidate molecules to choose from, tighter, more realistic filters can be applied and still enrich prediction outputs [1]. Computational drug discovery approaches generally fall into three categories: (i) physics-based simulations, (ii) machine learning (ML) surrogates that emulate those physics-based methods, and (iii) empirical ML models trained directly on experimental outcomes. Within these machine learning approaches, predictive (parametric scoring) models that assign scores to individual molecules through a direct forward pass generally provide the most reliable route to maximizing discovery yield. This stands in contrast to similarity-based retrieval methods, which rely on non-parametric lookups in molecular embedding space and therefore optimize for resemblance rather than novelty and discovery yield [2].

Figure 2: AstraZeneca Discovery Center Facility.
State-of-the-Art in AI Drug Discovery
Technologies such as Boltz-2 have generated significant attention for their ability to emulate physics-based accuracy [3]. However, despite improving computational efficiency relative to full FEP, Boltz-2 still demands immense resources and operates at a throughput of two million compounds per day on BioHive 2 (Figure 2) [3]. In contrast, AI-driven predictive models such as AtomNet (Atomwise) have achieved up to eight billion compounds per day throughput levels using a fully empirical ML forward-pass parametric scoring approach [4,5]. To our knowledge, however, no prior predictive screen has surpassed the hundred-billion scale.

Figure 1: Boltz-2 running on BioHive 2. Inference throughput of 20 seconds per molecule on one H100 GPU.
Problem with the State-of-the-Art
Current AI-driven drug discovery approaches suffer from two core misconceptions.
Problem 1: The “more training data is better” fallacy.
The prevailing belief that million to billion scale training datasets improve extrapolation is misleading. As data volume grows, redundancy of similar type chemistry increases, and rare-chemotype examples contribute almost nothing to the gradient updates. Models trained on this scale of data become finely tuned to the majority distribution, but blind to the rare, novel chemotypes that drive novel discovery.
Problem 2: Throughput neglect.
The industry’s fixation on large scale training has led to increasingly large models that achieve marginal accuracy gains at the expense of drastically reduced inference speed. In practice, many architectures cannot screen beyond a few million or billion molecules without prohibitive computational cost.
The consequence: lower novelty, shallow exploration, lower hit rates, and unnecessary attrition.
Our Solution and How We Did It:
Problem 1 Solution: Train on small, diverse datasets to improve extrapolation
GALILEO™ is engineered for extrapolation by prioritizing diversity over volume, training on orders-of-magnitude fewer, chemically varied data so rare chemotypes meaningfully influence learning and extrapolative power is preserved. During training and fine-tuning, a t-SNE guided data partitioning keeps scaffolds and local neighborhoods separated, explicitly pressuring the model to learn out-of-distribution structure rather than memorize nearby chemistries. Together, these choices make the model itself more creative, proposing candidates outside familiar neighborhoods. In prospective campaigns, GALILEO™ has surfaced hits chemically dissimilar to known active compounds, demonstrating genuine generalization rather than neighborhood memorization [6,7,8,9].
Problem 2 Solution: Lightweight, efficient architecture to enable extreme throughput
GALILEO™ uses a lightweight, efficient architecture built for high-throughput inference. Screening scales to orders of magnitude more candidates, enabling tighter filters (activity, novelty, ADMET, selectivity, patentability) that surface strong, lead-like compounds with greater chemical novelty. This Ultra-Large Virtual Screening (ULVS) capability unlocks a fundamentally broader exploration of chemical space, yielding higher novelty, improved hit rates, and dramatically reduced attrition [8].
Implementation: End-to-end on Google Cloud
Train on GPUs, infer on CPUs. Models are trained on GPUs for accuracy, then deployed for FP32 CPU inference to reach scale economically while preserving ranking fidelity.
Google Kubernetes Engine (GKE) schedules and autoscales thousands of containerized jobs for both library featurization and model inference (AMD EPYC fleets).
Google Cloud Artifact Registry stores versioned containers for featurization and inference, ensuring deterministic rollouts and easy rollback.
Cloud Storage holds input libraries, features, and all prediction outputs.
Operational principle. Workloads are containerized, reproducible, and embarrassingly parallel, so they scale cleanly.
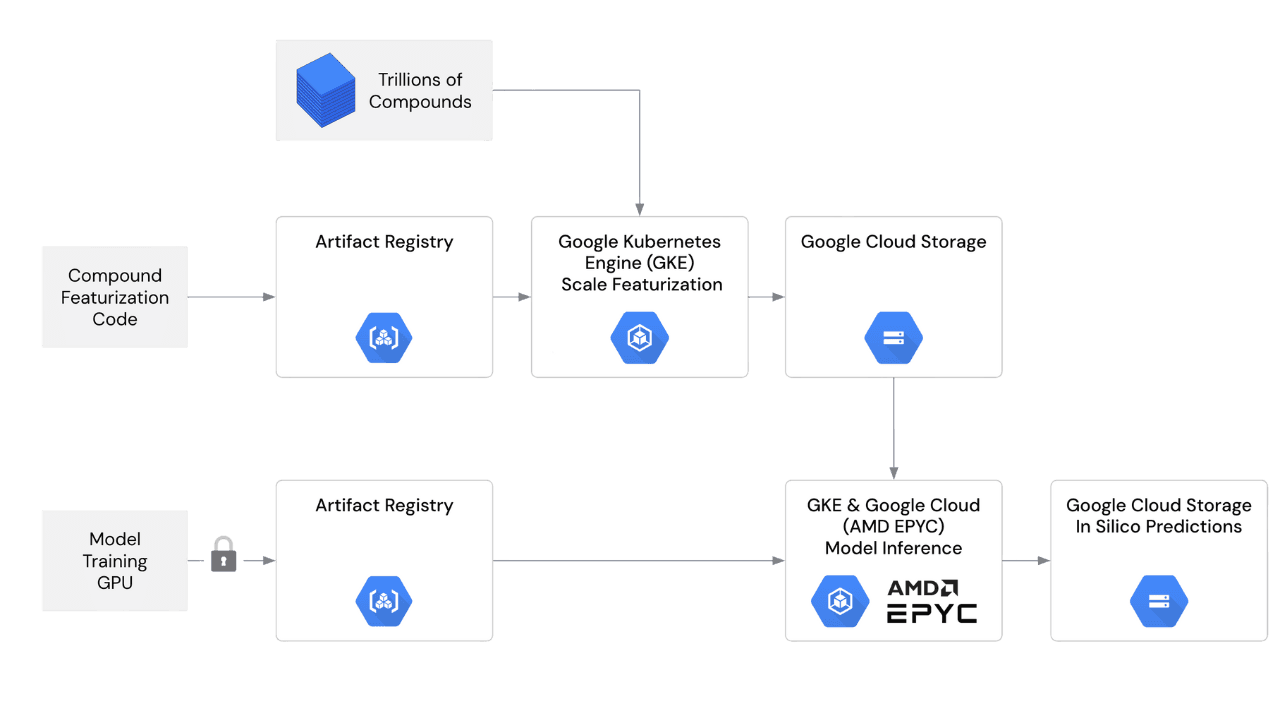
Figure 3: Model Medicines on Google Cloud. Google Cloud Artifact Registry contains images; GKE orchestrates containerized featurization and CPU inference on AMD EPYC fleets; Cloud Storage holds molecular features and prediction results.
Results - From Scale to Discovery
Hundred-Billion Scale Yields MDL-4102
GALILEO™ on Google Cloud achieved 325-billion molecule throughput, marking the first in silico campaign at hundred-billion scale, using a forward-pass parametric model that scores every molecule (Figure 4). This ULVS-scale screen powered our oncology program targeting BRD4, that led to the discovery of MDL‑4102, a next‑generation, first‑in‑class, selective BRD4 inhibitor and with no observable BRD2 nor BRD3 activity.

Figure 4: GALILEO™ running on Google Cloud. Running on 500 AMD EPYC CPUs, GALILEO achieved a record 325 billion compound throughput per day.
Chemical novelty.
MDL-4102 shows distinct, novel chemistry compared with BET inhibitors that have advanced into clinical testing. Tanimoto similarity analysis validates novelty to previously developed clinical compounds.
The BRD4 outcome illustrates GALILEO™’s ability to translate scale into targeted discovery, identifying novel candidates with therapeutic potential across a range of cancers.
Continuity with prior work.
This campaign builds on the Model Medicines Virology program (MDL-001) and our previously published NCE library traversal, where we reported exploration of ~53 trillion structures with potency, selectivity, and chemical novelty as designed [7,8]. Together, MDL-001 and MDL-4102 demonstrate the platform’s capacity to deliver category-defining therapeutics across distinct disease areas.
Where do we go from here?
Path forward.
The same FP32 CPU inference stack, containerized workflows, and GKE auto-scaling open the door to trillion-scale and even quadrillion-scale discovery, extending exploration to even sparser regions of chemical space while preserving ranking fidelity.
What Trillion-Scale Actually Takes
Across the industry, even leading AI drug-discovery players have in silico screening throughput constrained by cost and capacity: access to large A100/H100 fleets is limited and expensive, so campaigns typically cap at millions to low billions of molecules instead of reaching trillion-scale [3,4,5].
Capacity. Achieving trillion-molecule screening in 24 hours relies on embarrassingly parallel inference distributed across abundant AMD EPYC CPUs (Figure 5).
Why it matters. Abundant CPU availability plus containerized parallelism on GKE delivers predictable throughput and cost without GPU queue constraints.

Figure 5: Scaling the future of discovery. GALILEO™ is projected to reach trillion-scale screening by 2026 and quadrillion-scale by 2030.
Conclusion
State of the art today emphasizes ever-larger training data and models, yet most stacks cap practical screening at millions to low billions. Problem with the state of the art: (i) extrapolation suffers because billion scale training data drowns out rare chemotypes, pushing models to memorize local neighborhoods; (ii) throughput collapses as architectures scale up, making broad exploration cost-prohibitive. Approach to solving the problem: train for extrapolation with small, diverse data; engineer for throughput; run at ULVS scale on accessible cloud infrastructure. Results of the novel approach: GALILEO™ on Google Cloud achieved 325 billion molecule throughput, the first fully predictive in silico campaign at hundred-billion scale, using a forward-pass parametric model and FP32 CPU inference. This campaign delivered MDL-4102, a first-in-class, selective BRD4 inhibitor with no observable BRD2/BRD3 activity. MDL-4102 is chemically novel relative to clinical BET inhibitors (based on ECFP4 Tanimoto). By uniting models that generalize well with throughput engineering on Google Cloud, GALILEO™ establishes that novel therapeutic discovery scales with compute.
Liu, F., Mailhot, O., Glenn, I. S., Vigneron, S. F., Bassim, V., Xu, X., Fonseca-Valencia, K., Smith, M. S., Radchenko, D. S., Fraser, J. S., Moroz, Y. S., Irwin, J. J., & Shoichet, B. K. (2025). The impact of library size and scale of testing on virtual screening. Nature Chemical Biology, 21, 1039–1045. https://doi.org/10.1038/s41589-024-01797-w
Alnammi, M., Liu, S., Ericksen, S. S., Ananiev, G. E., Voter, A. F., Guo, S., Keck, J. L., Hoffmann, F. M., Wildman, S. A., & Gitter, A. (2023). Evaluating scalable supervised learning for synthesize-on-demand chemical libraries. Journal of Chemical Information and Modeling, 63(17), 5513–5528. https://doi.org/10.1021/acs.jcim.3c00912
Passaro, S., Corso, G., Wohlwend, J., Reveiz, M., Thaler, S., Ram Somnath, V., Getz, N., Portnoi, T., Roy, J., Stark, H., Kwabi-Addo, D., Beaini, D., Jaakkola, T., & Barzilay, R. (2025). Boltz-2: Towards accurate and efficient binding affinity prediction [Preprint]. bioRxiv. https://www.biorxiv.org/content/10.1101/2025.06.14.659707v1
Lin, Z., He, M., Mok, N. Y., Bricker, T., Lee, M., Wu, W., … & Hanson, R. M. (2024). AI is a viable alternative to high throughput screening: a 318-target study. Scientific Reports, 14, Article 7526. https://doi.org/10.1038/s41598-024-54655-z
N. Alarcon, “Atomwise Raises $123 Million to Accelerate AI Drug Discovery,” NVIDIA Technical Blog, Aug. 12, 2020. [Online]. Available: https://developer.nvidia.com/blog/atomwise-raises-123-million-to-speed-up-ai-drug-discovery/
Umansky, T. J., Woods, V. A., Russell, S. M., Smith, D. M., & Haders, D. J. (2024). ChemPrint: An AI-Driven Framework for Enhanced Drug Discovery [Preprint]. bioRxiv. https://doi.org/10.1101/2024.03.22.586314
Woods, V., Umansky, T., Russell, S. M., McGovern, B. L., Rosales, R., Rodriguez, M. L., van Bakel, H., Sordillo, E. M., Simon, V., García-Sastre, A., White, K. M., Brubaker, W. F., Smith, D., & Haders, D. (2025). MDL-001: An oral, safe, and well-tolerated broad-spectrum inhibitor of viral polymerases [Preprint]. bioRxiv. https://doi.org/10.1101/2025.01.13.632836
Umansky, T., Woods, V., Russell, S. M., Garvey, D. S., Smith, D. M., & Haders, D. (2025). GALILEO generatively expands chemical space and achieves one-shot identification of a library of novel, specific, next generation broad-spectrum antiviral compounds at high hit rates [Preprint]. bioRxiv. https://doi.org/10.1101/2025.01.17.633620
Umansky, T., Woods, V., Russell, S. M., & Haders, D. (2025). AmesNet: A deep learning model enhancing generalization in Ames mutagenicity prediction [Preprint]. bioRxiv. https://doi.org/10.1101/2025.03.20.644379

